

![]() Genetics, Mechanisms and Clinical Phenotypes of Arrhythmogenic Cardiomyopathy
Genetics, Mechanisms and Clinical Phenotypes of Arrhythmogenic Cardiomyopathy
Awardee: Frank I. Marcus, MD, Professor Emeritus, Medicine, past Chief, Division of Cardiology, UA College of Medicine – Tucson
Funding: NHLBI, NIH 1R01HL116906-01A1
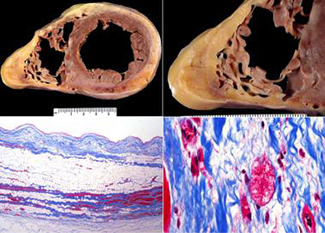 Arrhythmogenic ventricular cardiomyopathies (AVCs) are clinically and genetically heterogeneous heart muscle disorders characterized by frequent, often life-threatening arrhythmias that typically precede structural remodeling of the myocardium or clinical evidence of heart failure—more commonly referred to as sudden cardiac arrest.
Arrhythmogenic ventricular cardiomyopathies (AVCs) are clinically and genetically heterogeneous heart muscle disorders characterized by frequent, often life-threatening arrhythmias that typically precede structural remodeling of the myocardium or clinical evidence of heart failure—more commonly referred to as sudden cardiac arrest.
This sudden, unexpected loss of heart function often is fatal. Sudden cardiac arrest is not a heart attack (myocardial infarction), but can occur during one. It is the largest cause of natural death in the United States, causing about 325,000 adult deaths each year—and is considered responsible for half of all heart disease deaths. It’s estimated more than 7 million lives are lost each year to sudden cardiac deaths worldwide.
These AVCs are familial diseases but, despite progress in identifying various genetic causes, the majority of cases have no known specific cause or mechanism. AVCs include arrhythmogenic right ventricle (RV) cardiomyopathy (ARVC), arrhythmogenic left ventricle (LV) cardiomyopathy (ALVC), and a group of arrhythmogenic dilated cardiomyopathies (aDCM).
While these phenotypes may appear unrelated when compared by traditional clinicopathological criteria, recent evidence indicates that they may share common genetic causes and follow similar “final common pathways.” Mutations in genes encoding desmosomal proteins or desmosome-interacting proteins appear to play a major role in causing ARVC and ALVC. Recent evidence suggests some common genetic causes in ALVC and aDCM (and, therefore, ARVC) as well as mechanistic links between mutations in genes encoding Z-disk proteins and desmosomal disruption in aDCM. Despite progress in understanding the origin and progression of classical ARVC, many questions remain unresolved. The inability to identify causal genes in AVC patients represents a clear obstacle to individual patient risk-stratification. It also seriously limits the potential to identify preventive measures for at-risk or “silently” affected family members.
The goal of this Multicenter NIH funded research project is to enroll patients to our registry. Study the ongoing disease and its characteristics.
Specific aims for this study are to identify new genes causing ARVC, ALVC, and aDCM. To evaluate genotype-phenotype associations in patients with ARVC, to evaluate patients with suspected ALVC, aDCM; and to evaluate genotypephenotype associations in patients with ALVC and aDCM. To discover new biomarkers in AVCs and critically assess their value in improving the sensitivity and accuracy of diagnosis and to predict arrhythmias or other manifestations of disease.
ALSO SEE:
“40th Heart Rhythm Scientific Sessions Recognize Contributions of Dr. Frank Marcus to Understanding ARVC/D” | Posted May 15, 2019